Как указывалось выше, моделирование структуры начинается с формирования исходной конфигурации атомов, которая обычно выбирается случайным образом. Число атомов модели выбирается исходя из возможностей компьютера. Будем рассматривать простейший случай, когда конфигурация полностью характеризуется координатами точек, в которых расположены центры атомов. Эти точки при получении исходной конфигурации разбрасываются случайным образом внутри куба, сторона которого определяется плотностью моделируемого материала. Если начало координат выбрано в центре куба, то конфигурацию задают 3случайных чисел из равномерного распределения на интервале .
Искомая структура модели должна получаться автоматически в ходе работы алгоритма и определятся взаимодействием атомов. Парные потенциалы, описывающие взаимодействие всех типов атомов, входящих в химический состав задаются в модели исходно. Больше всего расчётов проводилось на основе наиболее простого предположения, что взаимодействие пары атомов и является ионным и описывается потенциалом Борна-Майера:
(2.3)
или степенным потенциалом:
.
(2.4)
В выражениях (2.3), (2.4) первое слагаемое описывает кулоновское взаимодействие, а второе – отталкивание заполненных атомных оболочек. и заряды ионов, – расстояние между ионами, и – числовые параметры, значения которых брались из литературы, или на основании сравнения расчетных ФРР с экспериментальными данными.
Таблица 2.2. Параметры потенциала Борна-Майера.
Энергия в таблице и на рис. 2.4 приводится в атомных единицах энергии (а.е.э.). 1 а.е.э.=4.35 10-11 эрг
Ионные пары
Aik, a.e.э
O-O
69.6
Si-O
67.6
Li-O
29.2
Na-O
51.2
Cs-O
Mg-O
Ba-O
Yb-O
Si-Si
42.4
Пример потенциала взаимодействия атомов кремния и кислорода изображён на рис. 2.4. Минимум потенциала расположен при несколько меньшем расстоянии между атомами, чем это наблюдается экспериментально для силикатных систем и воспроизводится для моделей. Дело в том, что на расстояние между кремнием и кислородом в тетраэдрах влияет кроме притяжения между кремнием и кислородом взаимное отталкивание атомов кислорода.
Энергия модели вычислялась путём суммирования всех парных взаимодействий (2.3, 2.4) каждого атома со всеми остальными атомами.
(2.5)
Исходная конфигурация модели, полученная путём случайного разброса атомов внутри куба, физически неестественна. В ней некоторые атомы расположены слишком близко друг от друга, отсутствует корреляция взаимного расположения атомов со знаком заряда: ионы одного знака могут оказаться соседями. Поэтому для исходной конфигурации значение энергии очень велико. Необходимо релаксировать модель, изменяя координаты составляющих её атомов так, чтобы получить наиболее энергетически выгодную конфигурацию, которую можно было бы рассматривать как равновесную. Чтобы уточнить это требование следует обратиться к статистической физике.
Рис 2.4 Парный потенциал взаимодействия (2.3) между атомами кремния и кислорода.
Согласно одному из её основных положений, система с фиксированными значениями объёма и числа частиц, будучи в равновесии с термостатом при температуре , может находиться в разных состояниях . При этом вероятность реализации конкретного состояния пропорциональна больцмановскому множителю:
,
(2.6)
где – энергия состояния , а – постоянная Больцмана. Полное выражение для содержит коэффициент, который здесь не приводится, чтобы не загромождать изложение, так как в пособии он не используется.
Распределение (2.6) называется «каноническим распределением», а ансамбль систем, определяемый этим распределением, – «каноническим ансамблем».
Задача алгоритма состоит в том, чтобы имитировать контакт модели с термостатом, имеющим заданное значение температуры . Термостат гораздо больше модели, и она не оказывает на него никакого влияния. Напротив, контакт с термостатом влияет на модель. Под действием этого влияния структура модели релаксирует и постепенно приобретает свойства реальной структуры. Релаксация проводится до тех пор, пока модель не придёт в равновесие с термостатом. После этого её конфигурации можно рассматривать как равновесные, соответствующие температуре . Исходную неравновесную конфигурацию, разумеется, температурой характеризовать нельзя.
Алгоритм, позволяющий имитировать перестройку структуры модели под влиянием контакта с термостатом, был предложен и опробован в работе Митрополиса и др., опубликованной в 1953 году [15]. В дальнейшем этот алгоритм получил чрезвычайно широкое распространение и фигурирует в современной литературе как «алгоритм Митрополиса».
Согласно этому алгоритму релаксация модели проводится путем последовательных случайных сдвигов всех атомов по одному с целью получения ансамбля конфигураций, которые можно было бы считать элементами канонического ансамбля при температуре Т.
Атом для сдвига выбирается случайным образом. Пусть оказалось, что он имеет номер «i». В результате сдвига координаты атома приобретают значения , , :
, ,
(2.7)
Здесь – значения координат выбранного атома до сдвига, – три случайных числа из интервала , максимальное изменение координаты. Сначала сдвиг рассматривается как «пробный». Решение о том принять его, или отвергнуть принимается в результате следующей процедуры.
Определяется величина изменения полной энергии модели, вызванная пробным сдвигом. Для этого необходимо вычислить энергию модели два раза – до сдвига и после сдвига. Условия принятия сдвига выглядят следующим образом:
если , сдвиг принимается (2.8)
если , сдвиг принимается с вероятностью
Второе условие реализуется согласно методике, описанной в разделе (1.3.1).
Если , то значение лежит в интервале [0,1]. Рассматриваются два возможных исхода: сдвиг принимается, сдвиг отвергается. Требуется, чтобы он принимался с вероятностью , а отвергался с вероятностью 1- . Чтобы это осуществить, используется очередное случайное число из интервала [0,1]. Если окажется, что , то сдвиг принимается. В противоположном случае – отвергается.
В работе Метрополиса с коллегами [16] показано, что в результате последовательности пробных сдвигов, которые принимаются при условии (2.8) модель постепенно переходит в равновесное состояние, соответствующее заданной температуре. Доказательство базируется на том, что последовательность конфигураций модели, получаемая при пробных сдвигах, обладает свойствами цепей Маркова.
Свидетельством того, что модель достигла равновесного состояния, могут служить следующие два теста.
Во-первых, энергия модели должна перестать монотонно убывать и испытывать только незначительные флуктуационные колебания около среднего значения.
Во-вторых, значения энергии, полученные для серии сдвигов после перехода в равновесное состояние, должны описываться каноническим распределением (2.6).
Чем выше температура, при которой проводится релаксация, тем быстрее достигается равновесное состояние. Но ФРР, для перегретого расплава, конечно, размыты и малоинтересны. Поэтому целесообразно проводить релаксацию при разных значениях температуры, последовательно ёе уменьшая. При этом исходное “физически бессмысленное” распределение атомов проще всего трансформируется в распределение, соответствующее очень высокой температуре.
После того, как устанавливается равновесие при заданной температуре (т.е. получающиеся конфигурации можно считать элементами канонического ансамбля) температура понижается и проводится релаксация при новом её значении. Получающиеся значения энергии модели при этом становятся меньше, а ФРР становятся более чёткими.
Периодические граничные условия.
Выше было сказано, что при построении исходного распределения атомы модели располагаются внутри куба со стороной . В дальнейшем при релаксации положения атомов внутри куба изменяются. При этом даже для моделей с наибольшим числом атомов, которое позволяют реализовать современные компьютеры, значительный процент атомов всегда оказывается вблизи от поверхности куба. Возникают две следующие проблемы:
(1) Характер воздействия окружения на выбранный атом сильно зависит от положения выбранного атома. Если выбранный атом расположен вблизи от центра куба, то остальные атомы действуют на него со всех сторон. Если выбранный атом располагается вблизи от границы, остальные атомы действуют на него с одной стороны. На атомы, расположенные в углу куба, действие остальных атомов сильно анизотропно.
(2) При пробном сдвиге атом может выйти из куба, в котором располагается модель?
Обе проблемы решаются путём введения следующих граничных условий.
Рис 2.5 Периодические граничные условия для двумерного случая.
Всё пространство заполняется одинаковыми кубами со стороной , так что вершины кубов образуют кубическую решётку. В каждом из кубов располагаются все атомы модели совершенно одинаковым способом. Каждый пробный сдвиг происходит одинаковым образом во всех кубах (рис. 2.5).
Если атом должен выйти при пробном сдвиге за границу куба, он попадает в такой же куб, а значит и в тот куб, из которого он уходит, но только с другой стороны.
Когда рассчитывается воздействие на выбранный атом остальных атомов модели, начало координат переносится в центр выбранного атома. При этом учитывается воздействие атомов, расположенных внутри куба со стороной , и с центром в новом начале координат (в атоме, воздействие на который рассчитывается). Таким образом, все атомы оказываются в одинаковом положении (в центре модели).
Моделирование структуры стёкол.
Одним из самых первых применений метода Монте-Карло было моделирование структуры жидкостей и расплавов (1953 г.). Однако, первые работы по моделированию методом Монте-Карло структуры стёкол (фторобериллатных и кварцевых) появились только в начале восьмидесятых годов [16,17,18].
Дело в том, что этот метод подразумевает возможность получения в результате релаксации термодинамически равновесного состояния. Такому условию удовлетворяет расплав стекла, при высокой температуре. При комнатной температуре классические стёкла находятся в неравновесном состоянии, и метод Монте-Карло к ним, строго говоря, не применим. Тем не менее, такие попытки были сделаны, что позволило получит ряд интересных результатов.
В частности, при моделировании изменений, происходящих в случае охлаждения стеклообразующих расплавов, были получены следующие результаты.
Зависимость энергии конфигурации от номера пробного сдвига отражала как монотонные изменения, связанные с релаксацией модели, так и флуктуационные колебания. О скорости релаксации давала представление величина, равная отношению понижения энергии конфигурации к числу пробных сдвигов DK, требовавшихся для этого понижения . Для определения по результатам модельного эксперимента необходимо было «сгладить» зависимость , аппроксимировав её монотонной кривой.
Величина в ходе релаксации убывала. Она уменьшалась практически до нуля за 104 сдвигов , в случае, если имитировалась структура при высокой температуре . В случае низких температур монотонное уменьшение энергии продолжало наблюдаться даже после серии сдвигов 107, и достичь равновесного состояния не удавалось. При этом значение скорости релаксации уменьшалось по сравнению с исходным в 105 раз, а изменение структурных параметров было трудно фиксировать. Авторы работ [16-18] рассматривали получающиеся в этом случае конфигурации как структурные модели стекла.
В пользу такой интерпретации говорили структурные характеристики модели, в качестве которых использовались парциальные ФРР, функции распределения углов, а также функции распределения, являющегося обобщением понятия «координационное число» (см. раздел 2.1). После длительной релаксации все названные характеристики находились в хорошем согласии с имеющимися данными дифракционных методов. В частности, на зависимостях хорошо выделялся пологий участок, соответствующий тетраэдрической координации стеклообразующих атомов – кремния по кислороду в стекле SiO2 и атомов бериллия по фтору в стекле BeF2 (рис. 2.6)
Автоматическое образование тетраэдров – неожиданный результат для чисто ионного взаимодействия. Следует подчеркнуть, что структура модели формировалась в ходе выполнения релаксационного алгоритма автоматически.
Рис 2.6 Зависимости обобщённого координационного числа от расстояния между атомами для для расплава и стекла BeF2. На рисунке введены следующие обозначения: - - FF, ++BeBe, + - BeF [17]
Аналолгичные результаты были получены и для других стеклообразователей.
Совсем иначе выглядят зависимости для других пар атомов: четкая координация для них отсутствует. Например, ион Eu3+ в фторобериллатном стекле является типичным модификатором. На зависимости от расстояния координационного числа европия по фтору пологий участок отсутствует (рис.2.7) Таким образом, в стекле имеется широкое распределение по значениям координационного числа для Eu3+ по фтору. Такой же результат был получен и для других редкоземельных ионов. Это объясняет особенности неоднородного уширения спектров редкоземельных ионов в стёклах [17].
Рис 2.7 Функции радиального распределения атомов вокруг европия в модели фторобериллатного стекла, активированного ионами Eu3+.
Первые Монте-Карло модели стекла состояли из нескольких сот атомов. В настоящее время строятся модели из нескольких тысяч атомов. Одно из достоинств модели состоит в том, что ёе можно применять для построения структур любого, сколь угодно сложного химического состава.

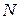 выбирается исходя из возможностей компьютера. Будем рассматривать простейший случай, когда конфигурация полностью характеризуется координатами точек, в которых расположены центры атомов. Эти точки при получении исходной конфигурации разбрасываются случайным образом внутри куба, сторона которого
выбирается исходя из возможностей компьютера. Будем рассматривать простейший случай, когда конфигурация полностью характеризуется координатами точек, в которых расположены центры атомов. Эти точки при получении исходной конфигурации разбрасываются случайным образом внутри куба, сторона которого  определяется плотностью моделируемого материала. Если начало координат выбрано в центре куба, то конфигурацию задают 3
определяется плотностью моделируемого материала. Если начало координат выбрано в центре куба, то конфигурацию задают 3 .
. и
и 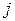 является ионным и описывается потенциалом Борна-Майера:
является ионным и описывается потенциалом Борна-Майера:
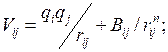
 .
.
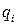 и
и  заряды ионов,
заряды ионов,  – расстояние между ионами,
– расстояние между ионами,  и
и 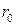 – числовые параметры, значения которых брались из литературы, или на основании сравнения расчетных ФРР с экспериментальными данными.
– числовые параметры, значения которых брались из литературы, или на основании сравнения расчетных ФРР с экспериментальными данными.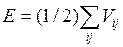
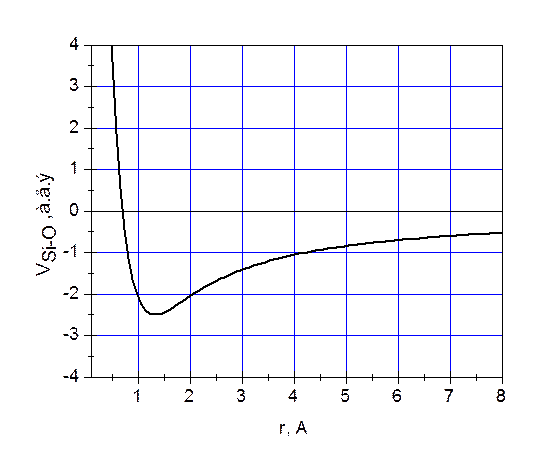
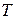 , может находиться в разных состояниях
, может находиться в разных состояниях 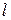 . При этом вероятность реализации конкретного состояния
. При этом вероятность реализации конкретного состояния 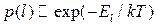 ,
,
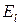 – энергия состояния
– энергия состояния 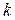 – постоянная Больцмана. Полное выражение для
– постоянная Больцмана. Полное выражение для 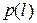 содержит коэффициент, который здесь не приводится, чтобы не загромождать изложение, так как в пособии он не используется.
содержит коэффициент, который здесь не приводится, чтобы не загромождать изложение, так как в пособии он не используется.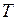 . Термостат гораздо больше модели, и она не оказывает на него никакого влияния. Напротив, контакт с термостатом влияет на модель. Под действием этого влияния структура модели релаксирует и постепенно приобретает свойства реальной структуры. Релаксация проводится до тех пор, пока модель не придёт в равновесие с термостатом. После этого её конфигурации можно рассматривать как равновесные, соответствующие температуре
. Термостат гораздо больше модели, и она не оказывает на него никакого влияния. Напротив, контакт с термостатом влияет на модель. Под действием этого влияния структура модели релаксирует и постепенно приобретает свойства реальной структуры. Релаксация проводится до тех пор, пока модель не придёт в равновесие с термостатом. После этого её конфигурации можно рассматривать как равновесные, соответствующие температуре 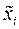 ,
, 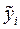 ,
, 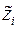 :
: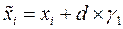 ,
, 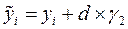 ,
, 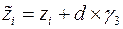
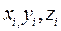 – значения координат выбранного атома до сдвига,
– значения координат выбранного атома до сдвига, 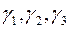 – три случайных числа из интервала
– три случайных числа из интервала 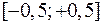 ,
,  максимальное изменение координаты. Сначала сдвиг рассматривается как «пробный». Решение о том принять его, или отвергнуть принимается в результате следующей процедуры.
максимальное изменение координаты. Сначала сдвиг рассматривается как «пробный». Решение о том принять его, или отвергнуть принимается в результате следующей процедуры. модели, вызванная пробным сдвигом. Для этого необходимо вычислить энергию модели два раза – до сдвига и после сдвига. Условия принятия сдвига выглядят следующим образом:
модели, вызванная пробным сдвигом. Для этого необходимо вычислить энергию модели два раза – до сдвига и после сдвига. Условия принятия сдвига выглядят следующим образом: , сдвиг принимается (2.8)
, сдвиг принимается (2.8)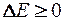 , сдвиг принимается с вероятностью
, сдвиг принимается с вероятностью 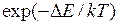
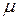 из интервала [0,1]. Если окажется, что
из интервала [0,1]. Если окажется, что 
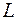 . В дальнейшем при релаксации положения атомов внутри куба изменяются. При этом даже для моделей с наибольшим числом атомов, которое позволяют реализовать современные компьютеры, значительный процент атомов всегда оказывается вблизи от поверхности куба. Возникают две следующие проблемы:
. В дальнейшем при релаксации положения атомов внутри куба изменяются. При этом даже для моделей с наибольшим числом атомов, которое позволяют реализовать современные компьютеры, значительный процент атомов всегда оказывается вблизи от поверхности куба. Возникают две следующие проблемы:
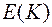 отражала как монотонные изменения, связанные с релаксацией модели, так и флуктуационные колебания. О скорости релаксации давала представление величина, равная отношению понижения энергии конфигурации
отражала как монотонные изменения, связанные с релаксацией модели, так и флуктуационные колебания. О скорости релаксации давала представление величина, равная отношению понижения энергии конфигурации 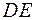 к числу пробных сдвигов DK, требовавшихся для этого понижения
к числу пробных сдвигов DK, требовавшихся для этого понижения  . Для определения
. Для определения 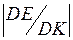 в ходе релаксации убывала. Она уменьшалась практически до нуля за
в ходе релаксации убывала. Она уменьшалась практически до нуля за  104 сдвигов , в случае, если имитировалась структура при высокой температуре
104 сдвигов , в случае, если имитировалась структура при высокой температуре 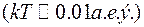 . В случае низких температур
. В случае низких температур  монотонное уменьшение энергии продолжало наблюдаться даже после серии сдвигов
монотонное уменьшение энергии продолжало наблюдаться даже после серии сдвигов 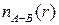 распределения, являющегося обобщением понятия «координационное число» (см. раздел 2.1). После длительной релаксации все названные характеристики находились в хорошем согласии с имеющимися данными дифракционных методов. В частности, на зависимостях
распределения, являющегося обобщением понятия «координационное число» (см. раздел 2.1). После длительной релаксации все названные характеристики находились в хорошем согласии с имеющимися данными дифракционных методов. В частности, на зависимостях 
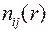 для для расплава и стекла BeF2. На рисунке введены следующие обозначения: - - FF, ++BeBe, + - BeF [17]
для для расплава и стекла BeF2. На рисунке введены следующие обозначения: - - FF, ++BeBe, + - BeF [17]
