Основной принцип различных алгоритмов расчета методом Монте – Карло основан на выборе случайных процессов в противоположность детерминированным алгоритмам. Расчеты методом Монте – Карло, в общем, подчиняются временной зависимости модели процесса, для которой изменение принимается не заранее заданным способом, а скорее случайным образом. Успех таких вычислительных алгоритмов зависит от быстрой и эффективной генерации последовательности случайных чисел (или чаще псевдослучайных чисел) во время моделирования.
В общем моделировании методами Монте – Карло самым подходящим является алгоритм, специально подобранный для данного случая. Разработанные ранее алгоритмы для моделирования макроскопических систем могут оказаться необоснованными для малых систем, представляющих интерес для нанотехнологии.
Методы Монте–Карло нашли разнообразное применение в вычислительной нанотехнологии. Достижения в нанотехнологии критически зависят т наличия быстрых и точных методов моделирования при конструировании и описании наноструктур. Имеются существенные проблемы при использовании методов Монте – Карло для предсказания особенностей свойств наносистем, если принимать во внимание тот факт, что куб с ребром 10 нанометров может содержать тысяча атомов и молекул.
В методах МД-моделирования особое значение имеет движение отдельных частиц (атомов и(или) молекул) внутри ансамбля из N атомов или молекул, составляющих изучаемую систему. Динамической теорией, используемой для получения уровня движения, является либо детерменированная динамика Ньютона, либо стохастическая динамика типа Ланжевена. При моделировании кристаллического или аморфного состояний необходимыми входными данными являются начальные координаты и скорости частиц, находящихся в первичной вычислительной ячейке объема V.
Для уменьшения времени расчета делается упрощающее предположение, что каждая частица взаимодействует только с ближайшими соседями, расположенными в то же ячейке, а также в транслируемых ячейках в пределах установленного радиуса обрыва потенциала. Тогда сиcтема из 3N сопряженных дифференциальных уравнений движения может быть решена различными численными конечно-разностными методами.
Атомы и молекулы твердого тела или кластера, за исключением водорода и гелия, у которых относительно небольшая масса, рассматриваются как объекты классической механики. Действительно, длина их тепловой волны при температуре Т, которая также называется длиной волны де Бройля, равна
где М выражается в единицах массы протона. Для водорода длина де Бройля имеет длину 1,0 Å, а для углерода она менее 0,3 Å (При Т = 0 К следует рассчитывать кинетическую энергию частицы вместо .
МД-моделирование – это метод , с помощью которого можно численно интегрировать классические уравнения движения и проследить траекторию движения атомов и молекул в некотором конечном временном интервале, не превышающем нано- или микросекунду. Из этих траекторий движения также можно получать данные о динамике атомов и молекул, визуально наблюдать за реакцией или рассчитывать механические и термодинамические свойства данной системы.
При расчете макроскопических свойств вещества с помощью молекулярной динамики, обычно предполагается, что в области с периодическими граничными условиями находится некоторое ограниченное число частиц исследуемого вещества. Периодические граничные условия означают, что если частица «вылетает» из области с одной стороны, то она возвращает обратно в эту же область с противоположной стороноы. Такой расчет помогает осуществить моделирование макроскопической системы с меньшими затратами компьютерного времени и избежать поверхностных эффектов, обусловленных конечным размером области. При этом однородная макроскопическая система путем трансляции элементарных ячеек заменяется областью с периодическими граничными условиями, в которой частицы передвигаются совершенно одинаковым образом во всех элементарных ячейках. Следовательно, важно полностью контролировать влияние дальнодействующих сил, оказываемое со сторны частиц, находящихся в соседних областях. Для МД-моделирования свойств наноразмерных веществ (таких, как открытый молекулярный кластер, ансамбль ограниченного числа частиц в замкнутом объеме и т.д.) не требуется никакого предположения о периодических граничных условиях.
Если вследствие ионной природы связей частицы оказываются заряженными, то между ними возникают дальнодействующие кулоновские взаимодействия. Стандартный способ решения этой проблемы – использование метода Эвальда для потенциала вида 1/R в трех измерениях[1].
Остальные взаимодействия, не относящиеся к ковалентным, обычно рассматриваются с помощью функций потенциалов. Но на практике эти потенциалы «обрываются», используя расстояния обрыва и сдвиг, как показано ниже:
,
где – функция эффективной потенциальной энергии взаимодействия.
Число операций на одну частицу, требуемое для вычисления силы межчастичного взаимодействия, в принципе, пропорционально общему числу парных взаимодействий в системе, если мы не учитываем многочастичные взвимодействия. Поэтому, в общем, на каждом временном шаге необходимо вычислять N(N-1)/2 сил взаимодействия. На практике из-за эффекта экранирования каждая частица испытывает воздействие только со стороны ее соседей, и для расчета всех сил взаимодействия требуется порядка cN вычислений, где с – среднее число ближайших соседей. Исключение составляют дальнодействующие силы, такие, как кулоновские. Однако многочисленные усилия совершаются для того, чтобы реализовать алгоритм сложности порядка N, в таком случае всегда полезно и необходимо иметь список соседей. Само собой составление списка соседей неизбежно приводит к большому числу вычислений. Список соседей содержит N(N-1)/2. Этот список необходимо сделать один раз в начале моделирования. Каждый раз после нескольких шагов по времени список необходимо обновлять, если моделируется жидкость или плотный газ, но эта задача требует сама по себе N вычислений, поскольку необходимо проверять только те частицы, которые, находясь во «второй, третьей…соседних оболочках», попадают в список соседей рассматриваемых частиц. По этой причине для создания списка соседей требуется большее расстояние обрыва, чем при подсчете сил взаимодействия.
Обычно для получения списка соседей за расстояние обрыва можно принять
,
где n – число шагов, после которых перечень соседей обновляется, и vmax – наиболее характерная (средняя) скорость частиц.
Для дальнодействующих кулоновских и гравитационных сил число расчетных шагов равно N(N-1)/2, которое в больших системах можно приближенно приравнять N2/2. При МД-моделировании большинства наносистем не требуется никакой аппроксимации, и можно приводить расчет с использованием всех N(N-1)/2 шагов, чтобы облегчить эту задачу, для больших систем можно использовать так называемый метод «частиц в ячейках».
При МД-моделировании наносистем каждую частицу внутри системы можно отождествить с точкой, затем расчетное пространство дискретезируется, и потенциальная энергия рассчитывается дискретно в соответствующих координатах сетки. Поскольку N – число узлов сетки, сложность задачи вычисления потенциала становится порядка N2/2. При очень большом числе частиц, когда время вычисления имеет значение, потенциальные энергии межмолекулярного взаимодействия сначала можно определить в Фурье-пространстве, чтобы воспользоваться более быстродействующим методом суммирования по Эвальду.
Если действующие силы известны во всех узлах сетки, интерполяцией можно найти их значения в местах фактического расположения частиц. Данное рассмотрение допускает, что потенциал, обусловленный кулоновским или иным другим дальнодействующим взаимодействием, является гладкой функцией, которая, в сущности, характеризует энергетическое поле.
Кроме того, можно приписать заряды каждому узлу сетки и полностью избежать суммирование по частицам I = 1…, N а затем решить уравнение Пуассона. Присвоение заряда сетке можно осуществить путем добавления объемных зарядов, распределяемых вокруг узлов сетки. В случае свободных граничных условий, как, напрмер, в гравитации можно решать уравнение Пуассона непосредственно в реальном пространстве. При использовании сетки размером М это эквивалентно решению системы линейных уравнений. В случае макроскопических и других больших систем, когда требуется задавать периодические граничные условия, для решения уравнения Пуассона можно использовать быстрое преобразование Фурье[2].
Допускается, что частицы, подобные точкам, двигаются согласно уравнению движения Ньютона
(3)
где Fi является результатом воздействия какого-нибудь внешнего поля, включая межчастичные взаимодействия. Эта простая форма уравнения Ньютона применима для изолированной системы, в которой выполняется закон сохранения полной энергии и импульса. С точки зрения статистической механики, уравнение (3) применимо для МД-моделирования микроканонического ансамбля, когда сохраняется полная энергия многочисленной системы. Позднее мы рассмотрим, как учесть влияние термостата (тепловой ванны) в уравнениях движения при моделировании канонических ансамблей.

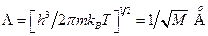
 .
.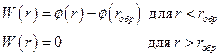 ,
, – функция эффективной потенциальной энергии взаимодействия.
– функция эффективной потенциальной энергии взаимодействия.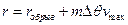 ,
,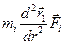 (3)
(3)